Sommige problemen lijken te complex om op te lossen. Een goed voorbeeld hiervan was de eiwitvouwing. Kunstmatige intelligentie blijkt dit ongelooflijk goed te kunnen. Hoe is dit mogelijk en wat heeft het voor consequenties?
Het grote vraagstuk achter eiwitvouwing is al in 1969 door Levinthal geformuleerd en staat bekend als de Levinthalparadox. Het aantal mogelijke opties aan structuren die een eiwit bij vouwing kan aannemen, is zo groot (bij 100 aminozuren: 10^300), dat het uitproberen van al deze mogelijke ‘conformaties’ oneindig lang zou duren. Dat is bijzonder, want in de praktijk vouwen eiwitten zich redelijk snel op.
Bijna al onze cellulaire processen worden door eiwitten geregeld. Elk eiwit bestaat uit een lange keten van aminozuren. Pas als je deze in de ruimte opvouwt krijgt een eiwit zijn functie, bijvoorbeeld als een enzym, dat een reactie kan versnellen, of als transcriptiefactor die aan DNA kan binden. Begrip van die ruimtelijke structuur helpt om te zien hoe het eiwit verandert na cellulaire signalen van buitenaf, zoals calciumbinding, hoe een transcriptiefactor aan DNA bindt om transcriptie te starten, of hoe een mutatie kan leiden tot verandering van functie. Een goed voorbeeld zijn de mutaties bij de omikronvariant van het coronavirus in het SARS-Cov2 Spike-eiwit, waardoor het virus beter aan de menselijke cellen kan binden. Ruimtelijke eiwitstructuren laten ook zien hoe een medicijn bindt aan een eiwit. Deze kennis is van groot belang bij de ontwikkeling van nieuwe medicijnen.
Om zo’n ruimtelijke eiwitstructuur te bepalen, zijn geavanceerde spectroscopische methodes nodig, waarmee je individuele atomen kunt onderscheiden. Dat kan met röntgenkristallografie, en sinds een paar jaar ook met elektronenmicroscopie. Bij de bacterie Escherichia coli kennen we ondertussen de ruimtelijke structuur van de meeste eiwitten, maar bij de mens geldt dat slechts voor een klein deel van de 20.000 menselijke eiwitten. Voor ieder individueel eiwit kostte het weken tot jaren om de structuur te ontrafelen. Vandaar dat er al lang gezocht wordt naar een snellere methode waarbij de structuur voorspeld wordt op basis van het de aminozuurvolgorde die is af te lezen uit het DNA. Daarbij helpt het dat de tegenwoordige DNA-databases over gegevens over het genoom van heel veel verschillende organismes beschikken.
Aan het voorspellen van de uiteindelijke structuur hebben onderzoekers al veertig jaar onderzoek gedaan. Ze worden hierbij geholpen door kennis van de evolutie. Om dezelfde functie te blijven vervullen, moet de structuur van een eiwit vrijwel gelijk blijven, terwijl de aminozuurvolgorde wel kan veranderen. Als meer dan 30% van de aminozuren van het eiwit hetzelfde is gebleven, komt de ruimtelijke structuur grotendeels overeen.
Doordat het DNA van veel organismes nu bekend is, is het mogelijk de aminozuurvolgorden van de varianten van een eiwit, de homologen, te vergelijken. Als de structuur van één homoloog bekend is, weet je al veel van andere homologen. Waarschijnlijk zitten verschillende aminozuren aan het oppervlak van een eiwit. Daar waar de grootte en lading niet uitmaken. Maar wanneer twee aminozuren binnen in het eiwit dicht bij elkaar zitten in de ruimtelijke structuur, veranderen ze vaak samen door de evolutie heen: als de een groter wordt, moet de ander kleiner zijn om daar plaats voor te maken. Of als de één van lading verandert moet de ander daarvoor compenseren. Dit valt onder het brede concept ‘co-evolutie’ en deze informatie is belangrijk in de structuurvoorspelling.
PROTEIN DATA BANK
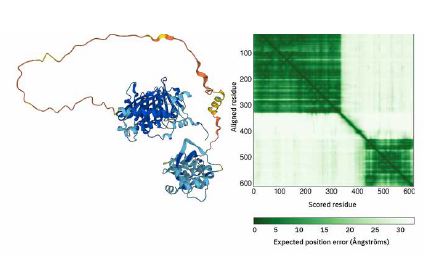
Op de webserver worden Alphafold voorspellingen op twee manieren weergegeven. In de afbeelding wordt links voor het DNA reparatie eiwit MutL met kleurtjes aangegeven waar de voorspelling goed is (blauw) en waar niet (oranje/geel). Daarnaast geeft Alphafold nog extra informatie: in de matrix rechts zie je dat de twee domeinen aan begin en einde van het eiwit intern wel interacties met andere aminozuren in dat domein maken (donkergroen), maar dat er tussen de twee domeinen waarschijnlijk weinig contacten zijn. Zo zie
je bijvoorbeeld geen donkergroen tusssen aminozuur 500 en aminozuur 200 in de matrix.
Onderzoekers hebben veel methodes ontwikkeld om structuren van eiwitten te voorspellen, maar het was lang moeilijk om te zien welk computerprogramma beter werkte. Door alle recente methodes te testen op experimentele eiwitstructuren, die wel bekend, maar nog niet gepubliceerd waren, werd het mogelijk deze methodes te vergelijken. Deze CASP-competitie vindt elke twee jaar plaats en heeft sterk geholpen om dit onderzoek te versnellen. Dit hielp om voorspellingen te verbeteren, maar het voorspellen van een nieuwe eiwitstructuur, waar geen varianten beschikbaar zijn bleef ver uit het zicht, tot de CASP-competitie van 2020, waar plotseling de methode van Google DeepMind, Alphafold, bijna perfecte voorspellingen gaf van het merendeel van de nieuwe structuren. Dit veroorzaakte een schokgolf van verbazing in het veld, zo’n gigantische stap voorwaarts maken we in de wetenschap niet vaak mee.
Hoe was dit mogelijk? Alphafold bouwt op veertig jaar onderzoek. Het gebruikt alle eerder bepaalde eiwitstructuren, die bewaard worden in een publiek toegankelijke database, de ‘protein data bank’(PDB, zie het kader). Het maakt ook gebruik van alle aminozuurvolgordes die bekend zijn dankzij de grote DNA sequencing-inspanningen. Hiermee kan de aminozuurvariatie en co-evolutie meegenomen worden. Dit zijn echte ‘big data’, bouwend op kennis uit de internationale wetenschap van de laatste veertig jaar. Er zijn dus veel data beschikbaar om het kunstmatige intelligentie algoritme te trainen.
Het Alphafold algoritme zelf is bijzonder innovatief. De DeepMind-onderzoekers hebben nieuwe manieren gevonden om de informatie uit de structuren en uit de aminozuurvolgordes aan elkaar te koppelen, in alle stappen van het proces. Daarnaast hebben ze een manier gevonden om de ruimtelijke structuur steeds vanuit dezelfde invalshoek te bekijken, dat wil zeggen ‘rotationeel invariant’. Bij een andere invalshoek zou het een andere structuur kunnen lijken terwijl het nog steeds dezelfde is.
De impact van Alphafold is enorm, en deze is versterkt doordat voorspellingen van ruimtelijke structuren van alle eiwitten waar een aminozuurvolgorde van bekend is, nu in een gemakkelijk toegankelijke database verzameld zijn (https://www.alphafold.ebi.ac.uk/). Dit helpt om snel naar nieuwe structuren te kijken, hoe het eiwit opvouwt en te zien waar bepaalde aminozuren kunnen zitten. Uiterst nuttige informatie die voor veel onderzoek van belang is.
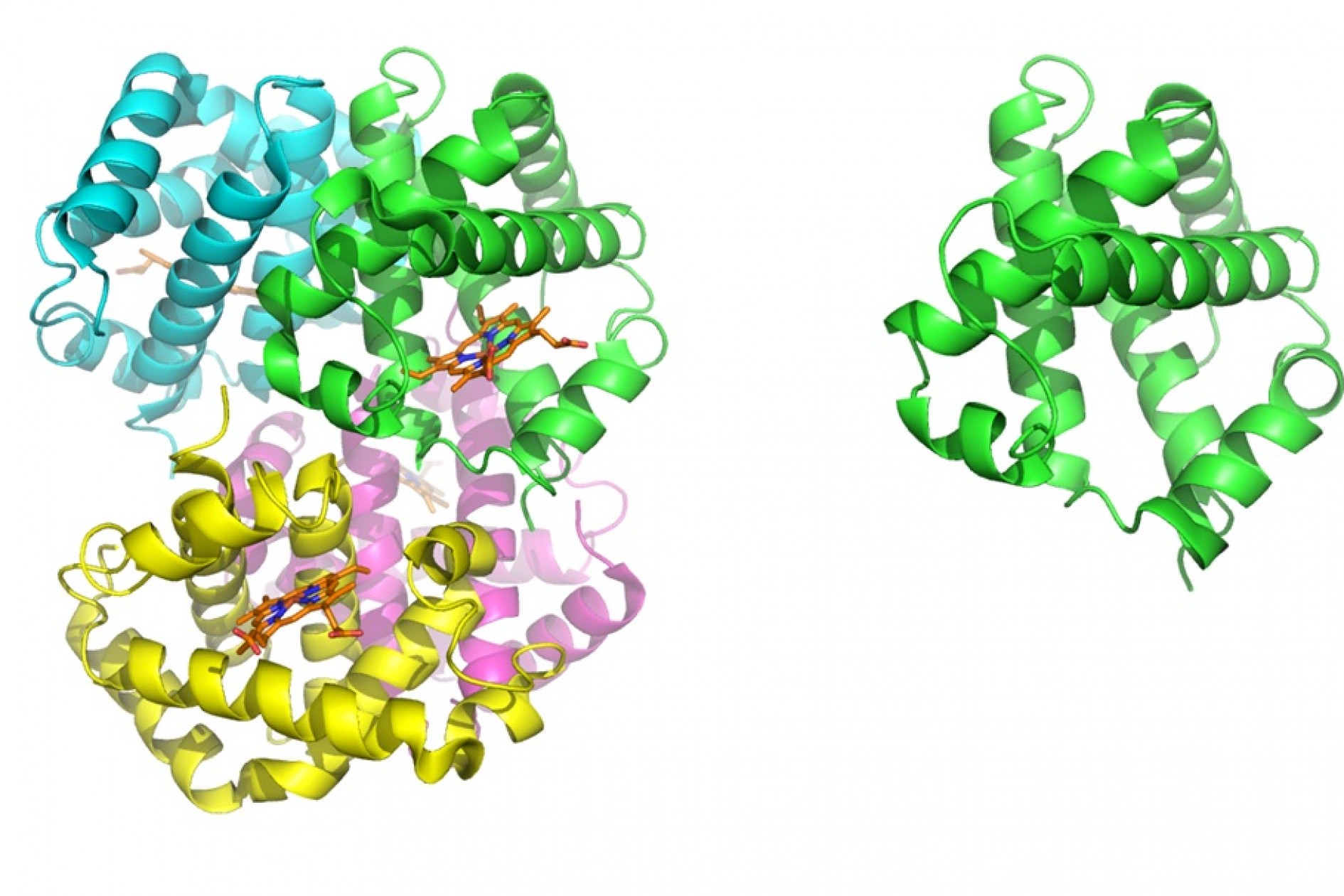
Alphafold heeft ook belangrijke beperkingen. Zo vertelt het niet hoe het vouwingsproces werkt, maar geeft alleen het eindstadium. Sterker nog, het kan een structuur voorspellen die in de praktijk niet voorkomt. Een goed voorbeeld is hemoglobine, het eiwit dat zuurstof vervoert. Hemoglobine bestaat uit vier eiwitketens, die elk een heem binden. Alphafold voorspelt de vouwing van één eiwitketen. De voorspelling is perfect, maar bij deze toestand ontbreken de cofactor heem en de andere eiwitketens. Om de voorspelling goed te gebruiken heb je dan ook aanvullende kennis nodig.
Alphafold kan ook niet voorspellen hoe een puntmutatie tot een structuurverandering leidt. Dat komt omdat Alphafold sterk bepaald wordt door de vergelijking tussen alle eiwitvarianten. Als zo’n mutatie niet opgetreden is, heeft Alphafold hier geen kennis over. Misschien is nog het belangrijkste probleem dat Alphafold voor elk eiwit slechts één enkele structuur voorspelt, terwijl de eiwitfunctie juist bepaald wordt door de veranderingen in conformatie. Als een eiwit ergens aan bindt, past het zich aan de partner aan. Als een eiwit in de vorm van een enzym een reactie versnelt, zijn kleine aanpassingen in het actief centrum van groot belang. Het zijn deze veranderingen die helpen te begrijpen hoe eiwitten werken, of die van belang zijn voor het ontwerpen van nieuwe medicijnen. Uiteindelijk zou je ook willen voorspellen hoe nieuwe medicijnen binden. Maar dat kan nog wel even duren: de gigantische databases die er voor de structuren en aminozuurvolgordes zijn, zijn hier niet beschikbaar. De variatie in moleculen en hoe ze binden is enorm, en de publiek toegankelijke data zijn relatief schaars.
Kortom, Alphafold biedt veel inzicht in nieuwe structuren en geeft snelle informatie over de structuur van veel eiwitten. Het versnelt nieuw onderzoek en geeft belangrijke inzichten. Maar zowel voor medicijnontwikkeling, als voor biologische kennis zijn experimentele data zeker nog essentieel.
